| ПОЗНАВАТЕЛЬНОЕ Сила воли ведет к действию, а позитивные действия формируют позитивное отношение Как определить диапазон голоса - ваш вокал Игровые автоматы с быстрым выводом Как самому избавиться от обидчивости Противоречивые взгляды на качества, присущие мужчинам Вкуснейший "Салат из свеклы с чесноком" Натюрморт и его изобразительные возможности Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д. Как научиться брать на себя ответственность Зачем нужны границы в отношениях с детьми? Световозвращающие элементы на детской одежде Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия Классификация ожирения по ИМТ (ВОЗ) Глава 3. Завет мужчины с женщиной
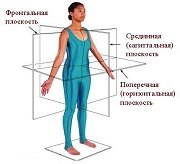
Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
| Пересмотренная классификация неходжкинских лимфом (REAL)
В-клеточные 1. НХЛ из предшественников В-клеток - пре-В-клеточный лимфобластный лейкоз/лимфома 2. В-клеточные опухоли из клеток с «периферическим» фенотипом (зрелых) а. В-клеточный ХЛЛ/мелкоклеточная лимфоцитарная лимфома б. Лимфоплазмоцитоидная лимфома/иммуноцитома в. Лимфома из мантийных клеток г. Лимфома из клеток фолликулярных центров, фолликулярная - из малых клеток, смешанная из малых и больших клеток, из больших клеток - диффузная, преимущественно из малых клеток д. В-клеточная НХЛ маргинальной зоны - экстранодальная (MALT ± моноцитоидная) - нодальная (± моноцитоидная) - селезеночная (± ворсинчатые лимфоциты) е. Волосатоклеточный лейкоз ж. Плазмоцитома/миелома з. Диффузная крупноклеточная В-клеточная лимфома подтип: первичная медиастинальная В-клеточная и. Лимфома Беркитта Т-клеточные 1. НХЛ из предшественников Т-клеток а. Т-лимфобластная лимфома/лейкоз 2. Т-клеточная НХЛ из клеток с «периферическим» фенотипом и NK-клеточные опухоли а. Т-клеточный ХЛЛ/пролимфоцитарный лейкоз б. Крупногранулярный лимфоцитарный лейкоз (Т-клеточный и NK-клеточный) в. Грибовидный микоз/синдром Сезари г. Т-клеточная лимфома с «периферическим» фенотипом - смешанная НХЛ из средних и больших клеток, из больших клеток, лимфоэпителиоидная д. Ангиоиммунобластная Т-клеточная лимфома (AILD) е. Ангиоцентрическая лимфома ж. Кишечная Т-клеточная лимфома (± энтеропатия) з. Т-клеточная лимфома/лейкоз взрослых (HTLV-1) и. Анапластическая крупноклеточная лимфома (CD30+), Т- и нулевого клеточного типа
Опухоли низкой степени злокачественностихарактеризуются медленным прогрессированием, длительной выживаемостью (годы), умеренной чувствительностью к химиотерапии и отсутствием возможности излечения при стандартной химиотерапии. В эту группу включены следующие варианты НХЛ (в соответствии с классификацией REAL): В-клеточные опухоли. 1. Фолликулярная НХЛ (I−II степени). 2. Диффузная лимфоцитарная НХЛ. 3. НХЛ маргинальной зоны: а) malt-экстраодалъная; б) моноцитовдная − нодальная; в) НХЛ селезенки. Т-клеточные НХЛ. Грибовидный микоз/синдром Сезари. Значительных различий в выживаемости внутри этой группы не выявлено. Высокоагрессивные лимфомы быстро прогрессируют, выживаемость составляет лишь месяцы, они умеренно и высокочувствительны к химиотерапии и могут быть излечены стандартными методами химиотерапии. Однако внутри этой группы НХЛ выявляются весьма значимые различия в продолжительности жизни: 5-летняя выживаемость колеблется от 78 % при анапластической крупноклеточной лимфоме до 14 % при лимфоме зоны мантии, в то время как при диффузной В-крупноклеточной лимфоме и фолликулярной лимфоме III степени этот показатель равен 38 и 68 % соответственно. В эту группу включены следующие варианты: В-клеточные опухоли. 1. Диффузная крупноклеточная НХЛ. 2. НХЛ Беркитта и беркиттоподобные опухоли. Т-клеточные НХЛ. 1. Лимфобластная лейкемия/лимфома. 2. Периферические Т-клеточные НХЛ. 3. Анапластическая крупноклеточная НХЛ. 4. Ангиоиммунобластная НХЛ. СТАДИИ ЛИМФОМ Стадия I: Вовлечение одной группы лимфатических узлов с любой стороны диафрагмы; непосредственное поражение ограниченной области или наличие одного экстранодального очага, являющегося единственным проявлением заболевания. Стадия II: Вовлечение 2-х и более групп лимфатических узлов по одну сторону диафрагмы; может вовлекаться селезенка, если некоторые группы лимфатических узлов расположены ниже диафрагмы. Стадия Ш: Вовлечение групп лимфатических узлов с обеих сторон диафрагмы, может вовлекаться селезенка. Стадия IV: Вовлечение экстранодальных участков, таких, как костный мозг или печень. А: Отсутствие симптомов интоксикации В: Лихорадка, ночные поты, потеря веса (более 10% массы тела) ± кожный зуд. Клиническая картина. НХЛ начинаются с появления одиночного опухолевого узла и распространяются путем лимфогенного и гематогенного метастазирования. Первичный опухолевый очаг может локализоваться в лимфатических узлах (нодальное поражение) или в других органах и тканях (экстранодальное поражение). Клинические проявления обусловлены расположением опухолевых очагов. Наиболее часто первыми проявлениями болезни бывает поражение лимфатических узлов (45-50 %); периферические лимфатические узлы вовлекаются в процесс значительно чаще (35-38 %), чем медиастинальные, забрюшинные и внутрибрюшные. Вначале лимфатические узлы плотны, безболезненны, не спаяны с кожей и подлежащими тканями. Позднее они образуют конгломераты, нередко довольно больших размеров. Изъязвление и образование свищей наблюдается не часто. При формировании свищевых ходов необходимо исключить туберкулез и актиномикоз. При поражении лимфатических узлов шеи и средостения может наблюдаться сдавление пищевода и трахеи, вызывающее дискомфорт при глотании пищи и кашель. Сдавление крупных сосудов грудной полости (наиболее часто в заднем медиастинальном пространстве) обусловливает застой в системе верхней полой вены, проявляющийся цианозом и отечностью верхней половины тела и лица с нарушениями дыхания и тахикардией. Лимфатические узлы брюшной полости и забрюшинного пространства, достигая значительных размеров или локализуясь в функционально ответственных зонах, могут обусловить развитие кишечной непроходимости, застой в портальной системе и нарушение лимфооттока из нижней половины тела (как следствие – развитие асцита, отеков нижних конечностей, половых органов), появление механической желтухи, нарушение мочеотделения. Опухолевые образования в носоглотке бугристые, бледно-розового цвета, быстро растут, могут прорастать решетчатый лабиринт, верхнечелюстную пазуху, глазницу, вызывая экзофтальм. Основными клиническими симптомами являются затруднение носового дыхания и снижение слуха. Глоточные миндалины могут значительно увеличиваться, а иногда (при двустороннем поражении) почти смыкаться между собой и быстро изъязвляться. При поражении молочной железы опухоль может определяться в виде диффузного уплотнения или довольно четкого опухолевого узла с прорастанием кожи и подлежащих тканей или без него. Симптом умбиликации (втяжение соска) не выражен. Поражение яичка начинается с отдельного участка, но быстро распространяется на весь орган, как правило, не вызывая спонтанных болей и пальпация безболезненна, но иногда опухоль прорастает кожу мошонки и распадается с образованием язвенной поверхности. При сочетании поражения яичка с регионарными (пахово-подвздошными) лимфатическими узлами отмечается отек мошонки. Лимфома кожи проявляется по-разному. Может иметь место постепенно растущий солитарный опухолевый узел в толще кожи и подкожной жировой клетчатке или появление вслед за маленьким внутрикожным узелком множества других узлов разных размеров. Обычно они плотны, безболезненны, могут располагаться группами и сливаться в плотные инфильтраты с бугристой поверхностью, склонные к изъязвлению. Цвет кожи над ними может быть не изменен или она приобретает темно-багровую окраску. У части больных первичные проявления протекают по типу дерматитов. Особенно характерная клиническая картина развивается при поражении ЦНС. Как правило, это является следствием гематогенного диссеминирования НХЛ и обнаруживается в среднем через 1,5 года от начала болезни преимущественно при НХЛ высокой степени злокачественности. Реже встречается изолированное первичное поражение ЦНС. Клиническая картина может быть мало выраженной и зависит от локализации опухоли: появление одиночного или реже множественных узловых образований в веществе головного мозга сопровождается неврологической симптоматикой поражения различных отделов головного мозга, а при вовлечении в процесс оболочек мозга наблюдается картина, напоминающая менингит (резкая головная боль, тошнота, рвота, положительные менингеальные симптомы). При расположении опухоли в желудочно-кишечном тракте не наблюдается специфических, присущих только НХЛ клинических признаков. Больные предъявляют жалобы, которые отмечаются при любой опухоли этой локализации (тошнота, ухудшение аппетита, снижение массы тела). Выраженность клинических проявлений заболевания обусловлена локализацией и формой роста опухоли. Наиболее часто поражаются желудок и тонкая кишка, как при изолированном, так и при сочетанием вовлечении их в процесс. Клинический вариант заболевания с исходным локализованным поражением экстранодальных органов и тканей определяется как первичная экстранодальная НХЛ. Подобная трактовка клинической ситуации возможна при выявлении единственного экстранодального опухолевого очага или в сочетании с последующим вовлечением в процесс регионарных лимфатических узлов (клинические стадии IE или IIЕ). Диагностика. Заболевание диагностируют на основании морфологического исследования опухолевого образования. Цитологическое исследование высокоинформативно. Следует широко выполнять его в амбулаторных условиях. Окончательным следует считать гистологическое исследование биоптата опухолевой ткани с иммунофенотипированием (Приложение 1). Цитологическая верификация допускается только в тех случаях, когда взятие материала для гистологического исследования cопряжено с высоким риском для жизни. Чаще всего выполняют биопсию периферических лимфатических узлов. Необходимо отдавать предпочтение биопсии длительно существующего лимфатического узла, расположенного в легко доступных анатомических областях (лучше шейно-надключичной области). Менее желательна биопсия паховых и апикальных аксиллярных лимфатических узлов). Целью иммунофенотипирования является определение В- и Т-клеточного происхождения опухоли и уровня нарушения дифференцировки лимфоидной клетки. Этот метод являются высокоинформативным компонентом комплексной диагностики НХЛ. Наиболее часто (более чем в 90 % случаев) НХЛ имеет В-клеточное происхождение, экспрессируя пан-В-клеточные антигены: CD19, CD20, CD22, обычно в сочетании с HLA/DR и молекулами поверхностных иммуноглобулинов. Наличие других В-клеточных антигенов (CD5, CD10, CD38, CD23 и др.) позволяет с наибольшей достоверностью установить В-клеточный вариант НХЛ, что лежит в основе выбора адекватной лечебной тактики. Для Т-клеточных опухолей характерно наличие CD4, CD7, CD8. Поскольку возможно расположение опухолей в любых органах и тканях, план обследования должен включать: полный физикальный осмотр с исследованием всех групп периферических лимфатических узлов, рентгенологическое исследование органов грудной клетки (для определения состояния медиастинальных лимфатических узлов и легочной ткани), ультразвуковое исследование печени, селезенки, внутрибрюшных и забрюшинных лимфатических узлов, фиброларингоскопию для выяснения состояния лимфа-тического аппарата кольца Пирогова-Вальдейера, рентгенологическое исследование желудка или, предпочтительно, гастроскопию с биопсией суспициозных участков слизистой оболочки, стернальную пункцию и трепанобиопсию подвздошной кости для исключения лейкемической трансформации и гематогенного очагового поражения костного мозга. Кроме того, нужно исследовать органы, со стороны которых больной испытывает дискомфорт. При особых клинических вариантах НХЛ применяют специфические дополнительные методы обследования: при поражении оболочек головного и спинного мозга (после осмотра неврологом) – люмбальную пункцию с определением клеточности цереброспинальной жидкости, и биохимического и цитологического исследования, при опухолевом поражении ЦНС – компьютерную томографию головного мозга и (или) уровня поражения спинного мозга, установленного при неврологическом осмотре; при первичном поражении одного из отделов желудочно-кишечного тракта – дополнительное исследование всех отделов желудочно-кишечного тракта. Диагностические критерии. Диагностику лимфом (лимфоцитом) облегчают следующие характерные особенности, присущие зрелоклеточным опухолям лимфатической системы: • очаговый характер лимфоидной пролиферации, поражение преимущественно какого-либо одного органа; • доброкачественное течение заболевания на протяжении 10-20 лет; • частое (около 25%) перерождение в лимфосаркому (А. И. Воробьев, 2000), которая очень чувствительна к химиотерапии и облучению и дает полную ремиссию; • невысокий лимфоцитов в периферической крови; • высокая частота секреции моноклонального иммуноглобулина (чаще IgM); • обнаружение в биоптатах лимфатических узлов пролиферации зрелоклеточных лимфоцитов. Прогноз. На основании тщательного изучения всех проявлений НХЛ было установлено, что наиболее значимыми факторами неблагоприятного прогноза являются: возраст старше 60 лет, повышение уровня ЛДГ (в 2 раза и более), общее состояние больного, соответствующее 2-4-й степени (ECOG), III − IV стадия болезни, наличие более одного экстранодального очага поражения, вовлечение костного мозга. Это было положено в основу Международного прогностического индекса – МПИ (IPI). Размеры опухолевых масс не включены в МПИ, хотя в ряде клинических ситуаций, бесспорно, играют отрицательную роль. На основании наличия одновременно того или иного количества неблагоприятных факторов прогноза различают четыре группы (степени) риска раннего прогрессирования болезни: низкую – отсутствие или присутствие лишь одного неблагоприятного признака, низкую/промежуточную – наличие двух факторов, промежуточную/высокую − присутствие трех и высокую − четырех неблагоприятных факторов Лечение. При НХЛ используют все виды противоопухолевой терапии. Основными факторами, влияющими на выбор терапевтической такта являются, распространенность процесса (I−II или III−IV клиническая стадия), морфологический вариант опухоли, первичная или преимущественная локализация опухолевого поражения, факторы прогноза. Единственным поистине объективным критерием эффективности терапии является подтверждение наступления молекулярной ремиссии – документированное использованием метода полимеразной цепной реакции, отсутствие моноклонального опухолевого клона и присущих ему молекулярных изменений. Эта оценка всё шире внедряется в мировую практику. Хирургическое лечение применяют с наименьшей частотой. Основным показанием к нему являются первичные одиночные опухоли желудочно-кишечного тракта. Спленэктомию выполняют редко, обычно с целью коррекции гематологических показателей при выраженном гиперспленизме (что часто сочетается со специфическим поражением селезенки). Лучевая терапия является высокоэффективным методом лечения НХЛ, может использоваться на всех этапах болезни, хотя возможности ее неодинаковы. В качестве самостоятельного метода лечения ее применяют редко, но успешно сочетают с другими лечебными воздействиями, особенно химиотерапией. Большое распространение получила комбинированная (химиолучевая) терапия в начальных (I–II А) стадиях НХЛ. Лечение в I–II стадии. Комбинированное лечение всегда следует начинать с химиотерапии, которая приводит к полным ремиссиям не менее чем у 70 % больных уже к моменту начала лучевого этапа лечения. Несмотря на это, лучевую терапию обязательно включают в комплекс лечебных мероприятий, так как она дает более стойкие местные лечебные результаты. Осуществляется комбинированное химиолучевое лечение в виде «сэндвича»: 2-3 цикла полихимиотерапии – лучевая терапия – 2-3 цикла полихимиотерапии. В I стадии НХЛ низкой степени злокачественности до и после лучевой терапии проводятся 2 цикла полихимиотерапии по схеме СОР (циклофосфан, винкристин, преднизолон). Альтернативным вариантом лечения больных этой группы могут быть 3 цикла полихимиотерапии с последующей лучевой терапией без дополнительной химиотерапии после облучения, если к моменту начала лучевой терапии была достигнута полная ремиссия. Больным с НХЛ высокой степени злокачественности в I стадии до начала лучевой терапии и после нее требуется провести по 3 цикла полихимиотерапии. В комбинацию химиопрепаратов должны быть включены антрациклины. Наиболее удобной и эффективной является схеме CHOP (циклофосфамид, доксорубицин, винкристин, преднизолон). Во IIА стадии всем больным независимо от степени злокачественности следует проводить 6 циклов полихимиотерапии: 3 до и 3 после лучевой терапии. Облучению подлежат только исходные зоны поражения (традиционный режим гамма-терапии до суммарной очаговой дозы 32–36 Гр). Профилактическое облучение всех групп лимфатических узлов нецелесообразно. Частота рецидивов составляет 18–30 %, но в течение первого года они возникают менее чем у 1/3 больных. Лечение в III–IV стадии. Комбинированная химиолучевая терапия в III стадии по результатам не превосходит обычную, а в IV стадии малоэффективна, так как облучение отдельных доступных зон носит паллиативный характер. Основным методом лечения распространенных форм НХЛ является химиотерапия, интенсивность которой зависит от степени злокачественности опухоли и клинических проявлений. НХЛ низкой степени злокачественности являются медленно прогрессирующими опухолями, которые характеризуются своеобразными клиническими особенностями. В последнее время все шире используется при лимфомах низкой степени злокачественности митоксантрон (новантрон) как компонент полихимиотерапии. В случаях благоприятного прогноза в качестве I линии терапии наибольшее распространение получила комбинация МСР (митоксантрон – 14 мг/м2 однократно в 1-й день, лейкеран – 6 мг/м2 с 1-го по 10-й день и преднизолон − 25 мг/м2 с 1-го по 10-й день). Этой комбинацией все чаще заменяют схему СОР. При неблагоприятном прогнозе на первом этапе индукционной терапии используют комбинацию MAP (митоксантрон, цитарабин, преднизолон), OPEN (винкристин, преднизолон, этопозид, новантрон), NOPP (новантрон, винкристин, натулан, преднизолон), MVLP (митоксантрон, тенипозид, лейкеран, преднизолон). Такое лечение проводят при всех вариантах НХЛ низкой степени злокачественности. НХЛ высокой степени злокачественности. Клиническими особенностями НХЛ высокой степени злокачественности являются быстрый рост опухолевых образований и склонность к раннему прогрессированию. Первой линией терапии диссеминированных НХЛ является схема СHОР, признанная «золотым стандартом». В настоящее время в рандомизированных исследованиях показана возможность повышения эффективности схемы СНОР у первичных больных: полная ремиссия достигается в 75-86% случаев, что превышает результативность стандартной схемы более чем на 20%. Это достигается интенсификацией режима двумя путями: сокращением интервала между циклами до 10-14 дней и (или) увеличением дозы доксорубицина до 70-80 мг/м2. Подобная интенсификация стала возможной лишь при обязательном использовании колониестимулирующих факторов – G-CSF, GМ-CSF (Г-КСФ, ГМ-КСФ). Разработана 2-х недельная модификация СНОР для лиц пожилого возраста (старше 70 лет) с заменой адриамицина форморубицином.
Хронический лимфолейкоз Хронический лимфолейкоз (ХЛЛ, лимфома из малых лимфоцитов или лимфоцитарная лимфома) – клональное лимфопролиферативное неопластическое заболевание, характеризующееся пролиферацией и увеличением в периферической крови количества зрелых лимфоцитов на фоне лимфоцитарной инфильтрации костного мозга, лимфатических узлов, селезёнки и других органов. Ежегодная заболеваемость хроническим лимфолейкозом в странах Европы и Северной Америки составляет 3–3,5 на 100 000 населения, а среди лиц старше 65 лет – до 20 на 100 000. Мужчины болеют чаще женщин (2:1). Диагностика. Предположение о наличиихронического лимфолейкоза может быть высказано на основании изменений картины крови – наличия лейкоцитоза с относительным и абсолютным лимфоцитозом. Считается, что лимфолейкоз должен быть заподозрен уже при абсолютном количестве лимфоцитов в крови более 5,0х109/л. Согласно современным критериям, установленным Международным рабочим совещанием в 1989г. для постановки диагноза хронического лимфолейкоза необходимо наличие трех признаков: 1) абсолютного количества лимфоцитов крови, превышающего 10,0•109/л; 2) обнаружения более 30 % лимфоцитов в костномозговом пунктате; 3) иммунологическе подтверждения наличия В-клеточного клона лейкемических лимфоцитов. При В-клеточном варианте заболевания на поверхности лейкемических лимфоцитов обнаруживается экспрессия В-клеточных антигенов CD19, CD20, CD24 и активационных антигенов CD5 и CD23. Иммунологическая характеристика В-клеточного ХЛЛ позволяет рассматривать его как опухоль, морфологическим субстратом которой являются первичноактивированные В-лимфоциты. Первичная активация (первая встреча с антигеном) В-лимфоцитов происходит в паракортикальной зоне лимфатического узла, поэтому, согласно последним классификациям лимфоидных опухолей (ВОЗ), В-клеточный ХЛЛ отнесен к опухолям периферических органов иммунной системы. Для В-лимфоцитов при ХЛЛ в отличие от нормальных В-лимфоцитов характерна также слабая экспрессия поверхностных иммуноглобулинов. Обычно на поверхности В-лимфоцитов при ХЛЛ обнаруживается IgM, нередко одновременно с IgD. В этом случае молекулы иммуноглобулинов обоих классов имеют одинаковые лёгкие цепи, идиотипы и вариабельные части, т.е. принадлежат к одному клону клеток. Как и нормальные В-лимфоциты, при В-ХЛЛ лимфоциты образуют розетки с эритроцитами мыши. Экспрессия антигена CD5, слабая экспрессия поверхностных иммуноглобулинов и розеткообразование с эритроцитами мышей считаются важнейшими иммунологическими характеристиками В-лимфоцитов при В-ХЛЛ. Число Т-лимфоцитов у больных В-ХЛЛ может быть нормальным, увеличенным или сниженным, но нередко нарушается соотношение Т-хелперов и Т-супрессоров и уменьшается число Т-киллеров. При многочисленных эпидемиологических исследованиях до сих пор не удалось оценить роль каких-либо мутагенных факторов (радиация, химические агенты или алкилирующие препараты и др.), как и роль вируса Эпштейна-Барра, в возникновении хронического лимфолейкоза. В то же время установлено, что неслучайные хромосомные аберрации, возникающие, как правило, под действием мутагенов, наблюдаются у большинства больных ХЛЛ. По данным VIII Международного рабочего совещания по ХЛЛ (1999), методом FISH их удается выявить почти у 90 % больных. Наиболее частой из структурных хромосомных аберраций является делеция длинного плеча хромосомы 13 (13q-). Она определяется у 55 % больных ХЛЛ. У 18 % больных встречается делеция длинного плеча хромосомы 11 (llq-), у 7 % – делеция короткого плеча хромосомы 17 (17р-), у 6 % – 6q-. В 4 % случаев обнаруживаются транслокации с участием хромосомы 14 (14q32). У 8-10 % – удлинение длинного плеча хромосомы 14 (14q+). Делеция llq- затрагивает место расположения гена ATM (ген атаксии – телеангиэктазии), который участвует в контроле цикла деления клетки. Выпадение или уменьшение продукции гена ATM может приводить к возникновению опухоли. Медиана выживаемости больных ХЛЛ с наличием llq- в 2-3 раза короче, чем у больных без этой аномалии. Делеция 17р - захватывает экзоны 5-9 короткого плеча хромосомы 17, где расположен ген р53 – супрессор опухолевого роста. Лишь 13q- не влияет на прогноз, остальные хромосомные аберрации оказывают неблагоприятное влияние на течение болезни (см. Приложение №2). Клиническая картина. Хронический лимфолейкоз начинается исподволь и в большинстве случаев на ранних этапах прогрессирует медленно. По мере развития заболевания постепенно нарастает лейкоцитоз, одновременно в лейкоцитарной формуле постепенно увеличивается количество лимфоцитов до 75-85-99 %. Преобладают зрелые формы, но, как правило, обнаруживается 5-10 % пролимфоцитов и нередко 1-2 % лимфобластов. Число эритроцитов, содержание гемоглобина и число тромбоцитов на ранних этапах болезни чаще нормальные, а при высоком лейкоцитозе и значительном лимфоцитозе обычно снижены либо за счет вытеснения здоровых ростков патологическими лимфоцитами, либо в связи с присоединением аутоиммунных осложнений. Для ХЛЛ характерно наличие в мазке крови теней Гумпрехта- Боткина – полуразрушенных при приготовлении мазка, размытых ядер лимфоцитов. При исследовании костномозгового пунктата больного ХЛЛ уже на ранних этапах болезни обнаруживается увеличение числа лимфоцитов до 40-50-60%. Гематологические изменения могут быть единственным проявлением заболевания в момент установления диагноза, но в большинстве слуаев даже при нерезко выраженных изменениях крови удается обнаружить небольшое увеличение лимфатических узлов. С течением времени у подавляющего числа больных наблюдается медленное генерализованное увеличение лимфатических узлов, имеющих тестоватую консистенцию и без присоединения инфекции совершенно безболезненных. При рентгенологическом исследовании в это время, как правило, обнаруживается увеличение лимфатических узлов средостения, а при ультразвуковом исследовании – увеличение узлов в брюшной полости и забрюшинном пространстве. Размеры узлов у разных больных и даже у одного больного в различных областях могут колебаться в широких пределах – от 1,5-2 до 10-15 см в диаметре. При гистологическом исследовании наблюдается стирание рисунка строения лимфатического узла, диффузная инфильтрация лимфоцитами и пролимфоцитами. Увеличение селезенки у большинства больных появляется позже, чем увеличение лимфатических узлов, и лишь у некоторых из них достигают огромных размеров. Еще позднее обычно увеличивается печень. Однако у отдельных больных увеличение селезенки и (или) печени выражено на протяжении всего заболевания. Темпы развития болезни, скорость увеличения количества лейкоцитов, размеров лимфатических узлов и селезёнки при ХЛЛ колеблются широких пределах. При хроническом лимфолейкозе в развитии болезни и ее клинических проявлениях, помимо лейкемической лимфоидной пролиферации, важную роль играют количественные и качественные изменения как патологических, так и нормальных лимфоцитов. Известно, что лейкемические В-лимфоциты при ХЛЛ мало чувствительны к антигеннным стимулам и продуцируют сниженное количество нормальных иммуноглобулинов. В то же время количество нормальных В-лимфоцитов резко уменьшено, что ведет к характерной для ХЛЛ гипогаммаглобулинемии, усугубляющейся по мере развития заболевания. Сниженное количество иммуноглобулинов, нередко являющееся отражением неспособности лейкемических В-лимфоцитов к антителообразованию, обычно коррелирует с частотой бактериальных инфекций. Кроме того, даже у больных с нормальным количеством Т-лимфоцитов и натуральных киллеров (NK-клетки) их функция резко снижена, что также вносит вклад в характерную для хронического лимфолейкоза склонность к повторным инфекциям иих тяжелому течению. Наиболее часто возникают инфекции дыхательных путей (бронхиты, пневмонии, плевриты), на долю которых приходится более половины инфекционных заболеваний при ХЛЛ. Пневмонии при ХЛЛ имеют склонность к распространению на оба легких. Следует подчеркнуть, что на начальных этапах развития пневмонии у больного ХЛЛ физикальные данные часто оказываются скудными, поэтому при возникновении лихорадки необходимо незамедлительно провести рентгенологическое исследование. Довольно часты также бактериальные или грибковые инфекции мочевыводящих путей, кожных покровов и мягких тканей с развитием абсцессов и флегмон, herpes zoster. Нередко наблюдается сочетание нескольких инфекционных очагов – пневмонии, инфекции мягких тканей, кожи, заканчивающееся картиной сепсиса. Другим важным следствием иммунных нарушений при ХЛЛ является возникновение аутоиммунных осложнений. Наиболее часто развивается аутоиммунная гемолитическая анемия, занимающая второе место (после инфекций) среди осложнений, характерных для ХЛЛ. Положительный антиглобулиновый тест (проба Кумбса) выявляется у 20-35 % больных, но аутоиммунная гемолитическая анемия развивается на протяжении заболевания у 10-25 %. Аутоиммунная тромбоцитопения встречается гораздо реже, примерно у 2-3 % больных. Однако она представляет большую опасность, чем аутоиммунная анемия, поскольку резкое снижение количества тромбоцитов нередко приводит к жизненно опасным кровотечениям. Реже возникает парциальная красноклеточная аплазия, характеризующаяся тяжелой анемией со снижением гематокрита до 25-20 % при отсутствии ретикулоцитов в крови и практически полном отсутствии эритрокариоцитов в костном мозге. Еще реже появляются антитела против нейтрофилов. Существуют две современные классификации ХЛЛ, отражающие стадийность течения болезни. Одна из них предложена в 1975 г. K.Rai и соавт. (табл. 5). Таблица 5. Классификация ХЛЛ по K.Rai и соавт.
Другая предложена в 1981 г. J.Binet и соавт.(табл. 6). Таблица 6.Классификация ХЛЛ по J.Binet и соавт.
В настоящее время именно эти 2 классификации используют для оценки и сопоставлении результатов терапии. Лечение. Важнейшим вопросом терапии ХЛЛ является вопрос о времени начала лечения, так как темпы развития болезни, скорость увеличения количества лейкоцитов, размеров лифатических узлов и селезенки при ХЛЛ колеблются в широких пределах. Больной не нуждается в лечении лишь до тех пор, пока стабильно сохраняется стадия 0–I no K.Rai или А по J.Binet. В настоящее время считаются общепринятыми и приводятся во всех руководствах следующие показания к незамедлительному началу цитостатической терапии: 1) наличие «общих» симптомов – усталость, потливость, снижение веса тела; 2) анемия или тромбоцитопения, обусловленная инфильтрацией костного мозга лейкемическими клетками; 3) аутоиммунная анемия или тромбоцитопения; 4) массивная лимфаденопатия или спленомегалия, создающие компрессионные проблемы; 5) большое число лимфоцитов в крови (выше 150,0•109/л); 6) удвоение абсолютного числа лимфоцитов в крови менее чем за 12 мес.; 7) увеличенная подверженность бактериальным инфекциям; 8) массивная лимфоцитарная инфильтрация костного мозга (более 80 % лимфоцитов в миелограмме); 9) наличие комплексных хромосомных аберраций; 10) продвинутая стадия болезни (стадия С по J.Binet, III–IV по K.Rai). Большинство гематологов начинают лечение больного уже при признаках стадии В по J.Binet или I–II по K.Rai, не дожидаясь появления симптомов декомпенсации. Современная эра в терапии ХЛЛ началась с серединыXX столетия. В 1949г. O.Pearson и соавт. впервые сообщили об уменьшении лимфоидной пролиферации при ХЛЛ под влиянием стероидных гормонов. Вторым важнейшим событием в развитии терапии ХЛЛ было появление алкилирующих препаратов. Первый из них – дериват азотистого иприта − хлорамбуцил (хлорбутин, лейкеран) синтезирован в 1953г. J.Everett и соавт, который с успехом использовался. Вслед за хлорамбуцилом был синтезирован ряд препаратов алкилирующего действия, апробированных при терапии ХЛЛ: циклофосфан, дегранол, дипин, фотрин, пафенцил и др., из которых только циклофосфан сохраняет значение до настоящего времени. При лечении первичных больных ХЛЛ наиболее предпочтительным препаратом в режиме монотерапии является флударабин.Однако у больных более преклонного возраста, имеющих неблагоприятный клинический статус и сопутствующие хронические воспалительные заболевания или рецидивирующую инфекцию, терапию следует начать с хлорамбуцила. Флударабин является в настоящее время наиболее активным агентом для лечения ХЛЛ. Вводится в/в капельно ежедневно в течение 5 дней каждые 28 дней из расчета 25 мг/м2. Больные, не отвечающие на 2-3 цикла лечения флударабином, как правило, должны быть переведены на альтернативные программы терапии. У пациентов с частичной ремиссией лечение флударабином может быть продолжено (1-2 цикла) до получения более значительного терапевтического эффекта, если при этом нет угрозы развития миелотоксичности или инфекционного осложнения. Как правило, терапевтический эффект наблюдается после 3-6 циклов терапии флударабином. Полные ремиссии достигаются примерно у 30% нелеченых больных ХЛЛ, при этом общее число положительных ответов превышает 70%. Стремление улучшить имеющиеся результаты привело к созданию в 70-80-х годах на базе алкилирующих препаратов (чаще всего циклофосфана) комбинированных лечебных схем. Наибольшее распространение получили схемы СОР, CHOP и CAP, ставшие золотым стандартом при лечении лимфом и апробированные на больших группах больных хроническим лимфолейкозом.
СОР: циклофосфан - 400 мг/м2 в сутки внутривенно или внутримышечно с 1-го по 5-й день винкристин - 1,4 мг/м2 (но не более 2 мг) внутривенно в 1-й день преднизолон - 60 мг/м2 внутрь с 1-го по 5-й день CHOP: циклофосфан - 750 мг/м2 внутривенно в 1-й день винкристин - 1,4 мг/м2 внутривенно в 1-й день адриамицин - 50 мг/м2 внутривенно в 1-й день преднизолон - 60 мг/м2 внутрь с 1-го по 5-й день САР: циклофосфан - 500 мг/м2 внутривенно в 1-й день адриамицин - 50 мг/м2 внутривенно в 1-й день преднизолон - 60 мг/м2 внутрь с 1-го по 5-й день Интервалы между циклами составляют 21−28 дней в зависимости от показателей крови. Дозы отдельных препаратов в этих схемах иногда варьируют.Разные авторы проводят от 6 до 12 циклов, стремясь получить максимальный эффект. Критерии эффективности терапии ХЛЛпредставлены втаблице 7. Таблица 7.Критерии оценки ответа на терапию ХЛЛ
Трансплантация костного мозга имеет ограничения при ХЛЛ (возраст и сопутствующие заболевания). Спленэктомия показана больным ХЛЛ с аутоиммунными анемией, тромбоцитопенией при низкой эффективности у них кортикостероидной терапии или больным с резко выраженной спленомегалией с клиникой компрессии внутренних органов и неэффективностью химиотерапии. Больные с низким риском агрессивного течения заболевания в течение многих лет не нуждаются в проведении цитостатического лечения и, как правило, умирают от причин не связанных с ХЛЛ; описаны спонтанные ремиссии у больных с ХЛЛ. У больныхс промежуточным риском течения заболевания также на протяжении длительного времени может отмечаться стабильность клинической картины, в то время как другая часть больных ХЛЛ погибает от ХЛЛ через несколько месяцев после верификации диагноза, несмотря на терапию. Смерть у больных лимфомами наступает чаще от инфекционных и геморрагических осложнений, развивающихся при прогрессии заболевания, а также как осложнений цитостатической терапии.
САРКОИДОЗ Саркоидоз – системный относительно доброкачественный гранулематоз неизвестной этиологии, характеризующийся скоплением активированных Т-лимфоцитов (CD4+) и мононуклеарных фагоцитов, образованием несекретирующих эпителиоидно-клеточных неказеифицированных гранулем. Преобладают внутригрудные проявления этого заболевания, однако описаны поражения всех органов и систем, кроме надпочечника. Актуальность знакомства с саркоидозом врачей общей практики и различных специальностей диктуется изменением организации оказания помощи этой группе больных в России. В течение нескольких десятилетий больные саркоидозом находятся под наблюдением фтизиатров (VIII группа учета), тогда как сотрудники ведущих институтов туберкулеза высказали мнение о том, что в сложившейся эпидемиологической ситуации целесообразно передать функцию наблюдения за больными саркоидозом в поликлинику по месту жительства (М.В.Шилова и др., 2001). Классификация.Согласно МКБ-10 саркоидоз отнесен к классу III «Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм» и подразделяется следующим образом. D86.0 Саркоидоз легких D86.1 Саркоидоз лимфатических узлов D86.2 Саркоидоз легких с саркоидозом лимфатических узлов D86.3 Саркоидоз кожи D86.8 Саркоидоз других уточненных и комбинированных локализаций В международной практике принято разделение внутригрудного саркоидоза на стадии, основанные на результатах лучевых исследований: Стадия III. Патология легочной паренхимы без лимфаденопатии. Стадия IV. Необратимый фиброз легких. В последние годы все чаще рекомендуют называть эти градации типами, а не стадиями саркоидоза, поскольку далеко не всегда существует их строгая хронологическая последовательность. Эпидемиология. Вновь выявленные случаи чаще всего регистрируются в возрасте 20–50 лет с пиком в 30–39 лет, 2/3 пациентов – женщины. Тем не менее существует саркоидоз детского возраста и саркоидоз у пожилых. В России, по данным С.Е.Борисова (1995), заболеваемость саркоидозом составляет 3 на 100 000 населения. Этиология. Согласно последним концепциям, причиной возникновения саркоидоза нельзя считать один фактор. Это сочетание генетических, экологических, инфекционных и иммунологических причин (ACCESS Research Group, 1999). В соответствии с Международным соглашением по саркоидозу (ATS/ERS/WASOG Statement on sarcoidosis, 1999) известно 3 этиологических фактора, приводящих к образованию гранулем: бактерии, грибы и паразиты; продукты растений и животных (пыльца, споры, белки); соединения металлов. Среди гранулематозов инфекционной природы выделяют 3 группы: инфекции, вызванные хорошо известными микроорганизмами; заболевания, вызванные микроорганизмами, определенными с помощью недавно установленных методов, которые не были выделены микробиологически; расстройства, для которых возбудитель точно не установлен, однако весьма вероятно предполагается. Саркоидоз пока остается в 3-й группе. Более вероятна не непосредственно этиологическая, а триггерная роль инфекции в патогенезе саркоидоза: постоянная антигенная стимуляция может вести к нарушению регуляции выработки цитокинов. Среди инфекционных гипотез наибольшее число публикаций посвящено микобактериям. В то же время потенциальными антигенными стимулами к развитию саркоидоза называют Chlamydia pneumoniae, Borrelia burgdorferi, Propionibacterium acnes, а также ряд вирусов, включая вирус простого герпеса и аденовирусы. Среди потенциальных факторов развития заболевания называют также многие факторы окружения человека (экология города, профессиональные факторы и др.), тогда как курение не приводит к учащению случаев развития саркоидоза. Патогенез. Гранулематозное воспаление является вариантом хронического воспаления, при котором в воспалительном клеточном инфильтрате преобладают производные моноцитов крови: макрофаги, эпителиоидные и гигантские многоядерные клетки, формирующие ограниченные компактные скопления. Частным случаем гранулематозного воспаления является эпителиоидно-клеточный гранулематоз, выделяют также варианты с диффузной инфильтрацией мононуклеарными фагоцитами и с макрофагальными гранулемами. Для развития гранулематоза необходима способность этиологического (повреждающего) агента вызывать в организме гиперчувствительность замедленного типа (А.А.Приймак и др., 1997). В основе патогенеза саркоидоза лежит накопление CD4+ T-лимфоцитов вследствие иммунного ответа Th-1 типа. Саркоидоз не сопровождается полной анергией, так как при известных признаках периферической анергии имеет место высокий уровень иммунологической активности макрофагов и лимфоцитов в местах развития патологического процесса. По неизвестной пока причине активированные макрофаги лимфоциты скапливаются в том или ином органе и продуцируют повышенное количество интерлейкинов-1 (IL-1), IL-2, IL-12, фактора некроза опухолей (TNF-альфа). TNF-альфа считают ключевым цитокином, участвующим в формировании гранулемы при саркоидозе. Кроме того, при саркоидозе доказана неконтролируемая выработка активированными альвеолярными макрофагами 1-альфа-гидроксилазы (в норме она вырабатывается в почках) с высокой аффинностью к 1,25-дигидроксикальциферолу, что приводит к эпизодам гиперкальциемии, которая может служить маркером активности процесса и иногда приводит к нефролитиазу. Опосредованную легочными альвеолярными макрофагами реакцию 1-альфа-гидроксилирования стимулирует гамма-интерферерон и угнетают глюкокортикоиды. Развитие гранулематозной реакции связывают также с нарушениями механизмов апоптоза (запрограммированной гибели клеток) иммунокомпентентных клеток. Клиническая диагностика. Клинически саркоидоз можно разделить на остротекущий и хронический. Деление это весьма условно, поскольку остротекущий саркоидоз сердца или ЦНС проявляется иначе, чем только внутригрудной саркоидоз, тем не менее оно применяется на практике. Для острого и подострого саркоидоза весьма характерен синдром Лефгрена – лихорадка, двусторонняя лимфаденопатия корней легких, полиартралгия и узловатая эритема. Возможны и неполные варианты этого синдрома – только эритема с лимфаденопатией, лимфаденопатия с артралгиями и т.д. Такие пациенты бывают выявлены при обращении к врачу, они предъявляют много жалоб, однако это является хорошим прогностическим признаком течения саркоидоза, особенно, если в эту фазу не применять глюкокортикоиды. Изолированная бессимптомная лимфаденопатия средостения (рентгенологический тип I), возникшая у лиц до 40 лет, также в 90% случаев протекает благоприятно и дает спонтанную ремиссию. Синдром Хеерфордта–Валденстрема диагностируют в случаях, когда у больного есть лихорадка, увеличение околоушных лимфатических узлов, передний увеит и паралич лицевого нерва (паралич Белла), но он встречается относительно редко. Хронически текущий, начинающийся незаметно, проявляющийся только нарастающей одышкой и слабостью процесс можно отнести к прогностически неблагоприятному. Как правило, это рентгенологические типы II и III, т.е. имеются изменения паренхимы легких. Одним из наиболее характерных симптомов саркоидоза любого типа течения является усталость, которая является одной из ведущих причин снижения качества жизни пациентов. Больные нередко отмечают только повышенную утомляемость при отсутствии каких-либо патологических признаков при осмотре и физикальном обследовании. Боль в грудной клетке является частым и необъяснимым симптомом при саркоидозе. Она имеет различную локализацию, не связана с актом дыхания, иногда по ощущениям находится на грани между болью и дискомфортом. Между наличием боли и выраженностью лимфаденопатии корреляции не выявлено. Не было связи боли с наличием и локализацией плевральных изменений, так же как и с другими изменениями в грудной клетке, выявляемыми на РКТ. При сборе анамнеза тщательно опрашивают в отношении ранее перенесенных артритов, «двусторонних прикорневых пневмоний», кожных проявлений и лимфаденопатии. Обязательно уточняют, не вызывали ли пациента на дообследование после прежних профилактических осмотров (флюорографии). Лучевая диагностика. Саркоидоз по характеру диагностического поиска является «диагнозом исключения», поскольку является не заразным и не злокачественным процессом. При первичной лучевой диагностике (профилактическое обследование) синдромы внутригрудной лимфаденопатии, инфильтрации, диссеминации, локальной тени или интерстициальных изменений требуют прежде всего исключения туберкулеза, опухолевого заболевания и неспецифического заболевания легких. В настоящее время основным методом лучевой диагностики диссеминированных процессов и внутригрудной лимфаденопатии является рентгеновская компьютерная томография высокого разрешения (РКТ), которая позволяет выявить ряд характерных скиалогических синдромов. Внутригрудная лимфаденопатия. На рентгенограмме выявляют расширение тени средостения за счет увеличенных лимфатических узлов (чаще бронхопульмональных, чем медиастинальных). Лимфаденопатия может быть обратимой. Именно I тип саркоидоза дает до 90% спонтанных ремиссий. В то же время в узлах могут происходить необратимые изменения вплоть до очаговой кальцинации или кальцинации по типу скорлупы ореха. Симптом диссеминации. Наиболее частым признаком саркоидоза II–III типа на РКТ являются мелкоочаговые тени. В легочной ткани выявляют множество рассеянных очаговых теней от милиарных до 0,7 см. Мелкие очаги, представляющие собой слияния эпителиоидных гранулем, коррелируют с перибронховаскулярными, перилобулярными и центрилобулярными изменениями в областях лимфатических сплетений. Чаще всего эти тени прилежат к костальной, междолевой или межсегментарной плевре и более тесно располагаются в аксиллярных зонах. При саркоидозе расположение очагов преимущественно «перилимфатическое», что характерно также для пневмокониоза и амилоидоза, но не для милиарного туберкулеза, при котором расположение очагов носит случайный характер. Периферическое расположение очагов, большое количество утолщенных междольковых перегородок, заметное утолщение междолевых щелей также указывают на саркоидоз. Симптом локальной тени. При пневмоническом рентгенологическом симптомокомплексе отмечены ложные «фокусы» – саркоидомы – скопления гранулем на ограниченном участке легкого в пределах субсегмента или сегмента в сочетании с инфильтративно-дистелектатическими уплотнениями. Локальные изменения при саркоидозе принято считать атипичными, в этих случаях саркоидоз распознают достаточно поздно. Фиброзные изменения при остром и подостром течении саркоидоза могут быть минимальными и формируются постепенно. При позднем выявлении хронически текущего саркоидоза фиброз может оказаться первым рентгенологическим признаком. Буллезно-дистрофические изменения. Инволюция диссеминированного процесса при саркоидозе сопровождается сетчато-тяжистой или петлистой деформацией легочного рисунка, а также симптомами обструкции – краевой эмфиземой, буллами, участками гиповентиляции легочной ткани. Изменения могут быть как односторонними, так и двусторонними. При первичном обследовании больных саркоидозом и особенно при физикально выявляемых изменениях со стороны печени, почек и селезенки целесообразно проведение РКТ органов брюшной полости и почек. Среди других методов визуализации при саркоидозе (особенно при первичном обследовании или прогрессировании процесса) показано проведение ультразвукового обследования печени, почек, сердца, щитовидной железы и органов малого таза. При таком активном подходе значительно чаще, чем было принято считать ранее, выявляют экстраторакальные проявления саркоидоза. Магнито-резонансное (ЯМР) обследование информативно при саркоидозе центральной нервной системы, печени, сердца. Поражения многих органов при саркоидозе подтверждают при сканировании с галлием и технецием. Функциональная диагностика. Исследование функции внешнего дыхания (запись кривой поток-объем форсированного выдоха) при саркоидозе на ранних стадиях выявляет обструктивные нарушения на уровне дистальной части дыхательного дерева (снижение мгновенной объемной скорости на уровне 75% от начала форсированного выдоха – МОС75). Следует заметить, что эти изменения могут быть частично обратимы при применении ингаляционных бронхолитиков. При прогрессировании процесса могут доминировать смешанные и рестриктивные нарушения со снижением жизненной емкости легких (ЖЕЛ). Достоверный диагноз рестрикции обеспечивает проведение общей бодиплетизмографии тела, которая выявляет снижение общей емкости легких (ОЕЛ). Обязательным компонентом первичного и ежегодного обследования больных саркоидозом является ЭКГ. В ряде стран в обязательный протокол первичного обследования входит мониторирование по Холтеру, поскольку именно тяжелые поражения сердца – аритмии и блокады – входят в перечень причин летальных исходов при саркоидозе. Экстраторакальные проявления саркоидозапредставлены в таблице 8. Таблица 8. Экстраторакальные проявления саркоидоза
Дифференциальная диагностика и критерии окончательного диагноза Вторым важнейшим этапом дифференциальной диагностики саркоидоза является исключение заболеваний опухолевой природы, к которым относят лимфомы, метастазы во внутригрудные лимфатические узлы, а также диссеминации опухолевой природы – милиарный канцероматоз, бронхиолоальвеолярный рак, множественные метастазы в легкие и др. Мировой опыт клинической медицины накопил множество патогномоничных клинических, лучевых и инструментальных косвенных диагностичеких признаков для каждого из этих заболеваний. Однако в каждом случае описаны исключения, атипичные случаи, диагностические заблуждения. Все это привело к тому, что «золотым стандартом» дифференциальной диагностики саркоидоза стала его гистологическая верификация. Материал может быть взят из различных органов – при биопсии периферических лимафтических узлов, кожи, селезенки, слюнных желез, печени и т.д. Чаще всего именно легкие, внутригрудные лимфатические узлы и плевра бывают объектами для взятия образца тканей. Материал получают при трансбронхиальной, видеоторакоскопической или открытой биопсии, во время медиастиноскопии, трансэзофагальной пункции, аспирационной биопсии тонкой иглой с цитологическим исследованием аспирата. Характерным патологическим признаком саркоидоза является дискретная, компактная, неказеифицированная эпителиоидно-клеточная гранулема. Она состоит из высокодифференцированных мононуклеарных (одноядерных) фагоцитов (эпителиоидных и гигантских клеток) и лимфоцитов. Гигантские клетки могут содержать цитоплазматические включения, такие как астероидные тельца и тельца Шаумана. Центральная часть гранулемы состоит преимущественно из CD4+лимфоцитов, тогда как CD8+лимфоциты представлены в периферической зоне. Среди инвазивных методов наиболее часто встречаются бронхоскопия и проведение трансбронхиальной биопсии, информативность которой варьирует от 30 до 70% в зависимости от квалификации специалиста и оснащенности кабинета. По степени инвазивности трансбронхиальная биопсия, проводимая под лучевым контролем, является оптимальным методом получения материала для гистологического подтверждения. Открытая биопсия является методом выбора, но в большинстве случаев должна уступить место трансторакальной видеоторакоскопической биопсии, как менее травматичному, но высокоинформативному методу. В соответствии с Международным соглашением по саркоидозу (ATS/ERS/WASOG Statement on sarcoidosis, 1999) морфологический диагноз саркоидоза легких основан на трех главных признаках: присутствие хорошо сформированной гранулемы и ободка из лимфоцитов и фибробластов по наружному ее краю; перилимфатическое интерстициальное распределение гранулем (именно это делает трансбронхиальную биопсию чувствительным диагностическим методом) и исключение других причин образования гранулем. Проба Квейма–Зильцбаха. В 1941 г. норвежский дерматолог Ансгар Квейм обнаружил, что внутрикожное введение ткани лимфатического узла, пораженного саркоидозом, вызывает образование папулы у 12 из 13 больных саркоидозом. Луис Зильцбах усовершенствовал этот тест, используя суспензию селезенки, подтвердил его специфичность и организовал его проведение, как международное исследовние. Тест был назван пробой Квейма–Зильцбаха (Kveim– Siltzbach). В настоящее время эта проба представляет собой внутрикожное введение пастеризованной суспензии селезенки, пораженной саркоидозом. В месте введения постепенно появляется папула, которая достигает максимального размера (3-8 см) через 4-6 нед. Биопсия этой папулы в 70–90% случаев у больных саркоидозом позволяет обнаружить изменения, подобные саркоидозу. Развитие гранулем у больных саркоидозом |